Article Text

Abstract
Introduction Overactive bladder (OAB) is a condition characterised by urinary urgency, often accompanied by frequency and nocturia. Antimuscarinics and β3 receptor agonists are first-line therapies that improve urinary symptoms and the quality of life. For insufficient antimuscarinic response, options include dose increase, switching medications or combination therapy. However, evidence for these strategies, especially combinations with vibegron, is limited and needs further study.
Methods and analysis The study is designed as a randomised, open-label, parallel-group, multicentre trial conducted in Japan. A total of 110 patients with OAB who met the OAB criteria and did not respond adequately to the initial 4-week antimuscarinic treatment will be randomised in a 1:1 ratio into two groups: an add-on group in which vibegron 50 mg/day is added to the current antimuscarinic drug and a switch group in which the current antimuscarinics are discontinued and replaced with vibegron 50 mg/day. The primary endpoint is the intergroup comparison of changes in daily urinary frequency between the add-on group and the switch group at 12 weeks after the initiation of protocol treatment. The primary analysis aims to confirm the non-inferiority of the switch group compared with the add-on group using a Bayesian mixed model for repeated measures. Non-inferiority will be confirmed if the posterior probability that the difference in the change in urinary frequency at 12 weeks between the two groups falls within the non-inferiority margin of one-time is 80% or greater.
Ethics and dissemination The trial has been reviewed and approved by the Institute of Science Tokyo Certified Clinical Research Review Board (approval number: NR2024-001). Participants will provide informed consent to participate before taking part in the study. Results will be reported in a separate publication.
Trial registration number Japan Registry of Clinical Trials (jRCT) (jRCTs031240134).
- UROLOGY
- Bladder disorders
- Urinary incontinences
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
STRENGTHS AND LIMITATIONS OF THIS STUDY
The study is designed as a randomised, open-label, parallel-group, multicentre trial conducted to assess the non-inferiority of switching to vibegron following antimuscarinic therapy in patients with overactive bladder (OAB).
The adoption of Bayesian statistics, an innovative approach in the ADVISR trial, is expected to provide deeper insights into OAB treatment and more precise treatment effect evaluation.
This open-label randomised controlled trial with unblinded participants and therapists has an inherent risk of selection bias.
Introduction
Overactive bladder (OAB) is a condition characterised by urinary urgency, often accompanied by frequency and nocturia, with or without urgency urinary incontinence (UUI). Various factors can cause OAB, including age-related changes in bladder function, weakening of the muscles supporting the bladder and urethra, benign prostatic hyperplasia and the aftereffects of cerebral haemorrhage or stroke. OAB is diagnosed when urinary urgency occurs at least once a week and the total OAB Symptom Score (OABSS) is 3 or higher. A comprehensive Japanese epidemiological study of patients aged 40 and above revealed that, in 2012, the prevalence of OAB symptoms and urinary urgency was 14.1% for both.1 This study defined OAB symptoms as urinating eight or more times daily and experiencing urgency at least once a week. Based on this research using Japan’s 2012 population data, an estimated 10.4 million individuals were affected by OAB symptoms.1
Antimuscarinics that inhibit bladder contractions have been the main pharmacological therapy for OAB, but recently, β3 receptor agonists have emerged as medications that relax bladder muscle tone. The 2024 guidelines from the American Urological Association (AUA) and the Society of Urodynamics, Female Pelvic Medicine & Urogenital Reconstruction (SUFU) recommend that clinicians offer antimuscarinics or β3 receptor agonists as evidence-level grade A to improve urinary urgency, frequency and/or urgency incontinence in patients with OAB.2 Clinical studies have also demonstrated that OAB agents significantly improve other outcomes of interest, including overall and disease-specific quality of life (QOL), treatment satisfaction and work productivity.2
When antimuscarinics are insufficiently effective, dose increase, switching to other antimuscarinics, changing to β3 receptor agonists or introducing combination therapy should be considered. There is limited evidence that patients who do not respond to first-line antimuscarinics will respond to higher doses or different antimuscarinics.3 In Japanese guidelines, switching from antimuscarinics to β3 receptor agonists is expected to reduce antimuscarinic-specific adverse effects such as dry mouth and constipation, and is recommended as grade B.4 However, the efficacy of this switch in improving symptoms in patients who do not respond adequately to antimuscarinics is not yet well established and remains a grade C1 recommendation.4 For patients who do not respond adequately to antimuscarinics, a combination therapy with β3 receptor agonists is also an option. The 2024 AUA/SUFU guidelines recommend the combination of antimuscarinics and β3 receptor agonists as a grade B recommendation for patients with OAB who do not achieve sufficient improvement with monotherapy.2 In Japan, only the combination of antimuscarinic solifenacin succinate and β3 receptor agonist mirabegron is recommended for this combination therapy.4 Several clinical trials have shown that this combination therapy significantly reduces daily urination frequency and urinary incontinence episodes in patients who did not respond adequately to the antimuscarinic drug solifenacin succinate alone.5–10 However, evidence for the combination of other antimuscarinics and mirabegron is currently limited.
A Phase IIb clinical trial demonstrated that combining β3 receptor agonists vibegron 100 mg/day with antimuscarinic drug tolterodine 4 mg/day significantly reduced both the mean daily urination frequency and urinary urgency episodes compared with tolterodine alone.11 However, the efficacy of combining antimuscarinics with the standard Japanese vibegron dose (50 mg/day) remains inadequately evaluated. In a Phase III long-term clinical trial conducted in Japan to evaluate the safety and efficacy of vibegron 50 mg/day for 52 weeks, antimuscarinics were concomitantly administered in some cases, but the efficacy and safety of these drugs have not been fully evaluated.12 In other research, Tachikawa et al reported real-world data on the efficacy and safety of vibegron, but cases of concomitant use with antimuscarinics were excluded from the analysis.13 While numerous studies have reported on the efficacy and safety of mirabegron in combination with antimuscarinics for secondary OAB treatment, research on vibegron remains scarce, resulting in a significant evidence gap.5–10
In this Phase II randomised trial—ADd-on or switch to Vibegron in patients with OAB InSufficiently Responding to initial 4-week antimuscarinics (ADVISR trial)—we will assess vibegron’s efficacy and safety in two scenarios: as an adjunct to antimuscarinics and as their replacement.
Methods and analysis
Trial design
This study is a randomised, open-label, parallel-group, multicentre trial conducted in Japan. Participating institutions include the Institute of Science Tokyo, Tokyo Metropolitan Tama-Nambu Chiiki Hospital, Japanese Red Cross Oomori Hospital, Showa General Hospital, Kohnodai Hospital, National Center for Global Health and Medicine, Saitama Red Cross Hospital, Soka Municipal Hospital, Tsuchiura Kyodo General Hospital, Tokyo Metropolitan Ohtsuka Hospital, Tokyo Metropolitan Cancer and Infectious Diseases Center Komagome Hospital and the JA Toride Medical Center.
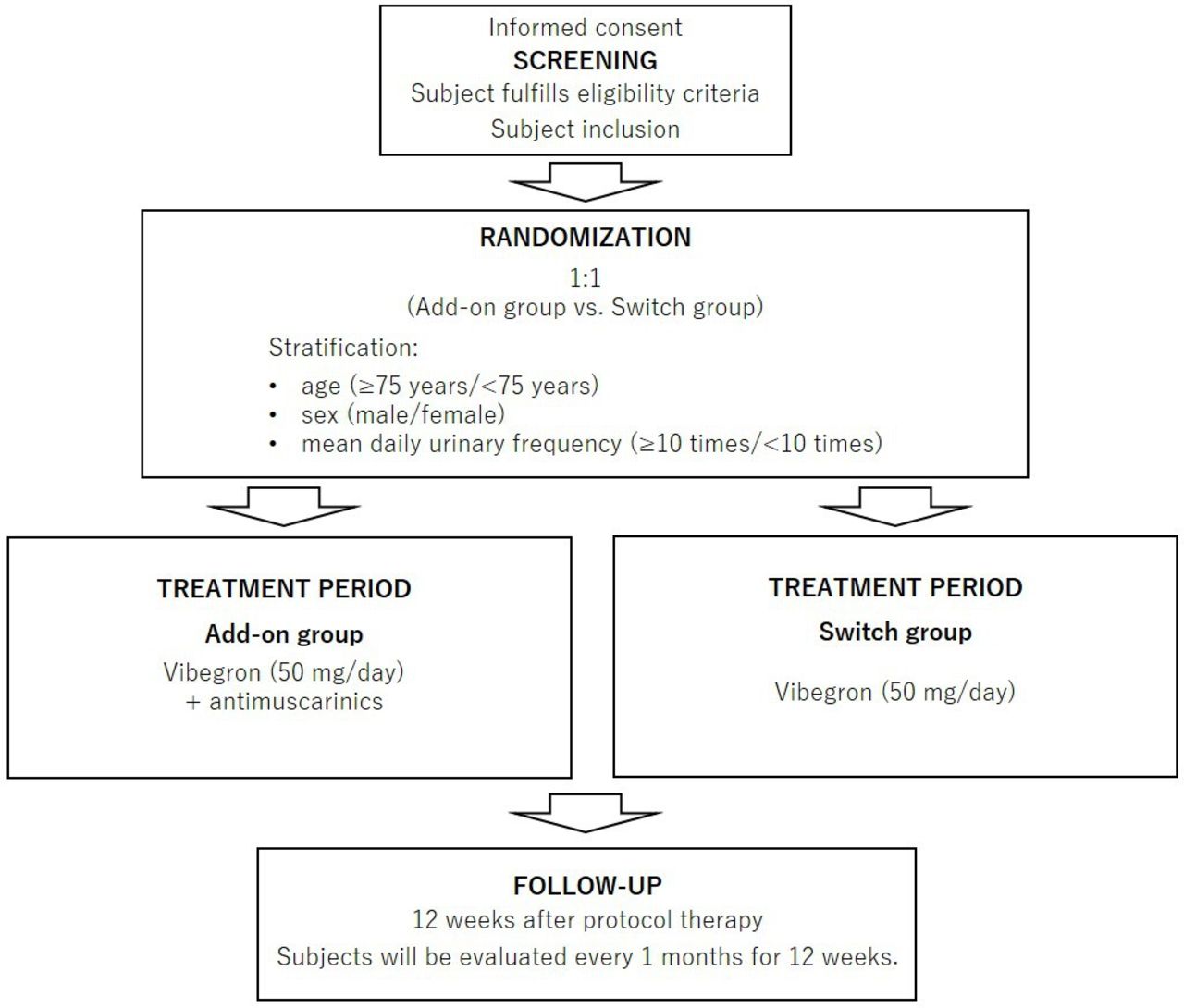
Patients with OAB who did not respond adequately to the initial 4-week antimuscarinic treatment will be randomised in a 1:1 ratio into two groups: an add-on group in which vibegron 50 mg/day is added to the antimuscarinic drug they were taking, and a switch group in which the antimuscarinic they were taking is discontinued and replaced with vibegron 50 mg/day. The definition of inadequate response was to meet the OAB criteria after initial the 4-week antimuscarinic treatment. A dynamic allocation method (ie, minimisation) with a random element in treatment assignment via an Interactive Web Response Systems is used with the following stratification factors: age (≥75 years/<75 years), sex (men/women) and average daily urinary frequency (≥10 times/<10 times). The primary endpoint is the intergroup comparison of changes in daily urinary frequency between the add-on group and the switch group at 12 weeks after the initiation of protocol treatment.
The study protocol follows the Standard Protocol Items: Recommendations for Interventional Trials (see 0nline supplemental additional file 1, 2). This trial has been reviewed and approved by the Institute of Science Tokyo Certified Clinical Research Review Board (approval number: NR2024-001) and was registered in the Japan Registry of Clinical Trials (jRCT) on 3 June 2024 (registration number: jRCTs031240134).14
Participants
The following inclusion criteria will be used for this trial:
Patients are aged 18 years or older at the time of enrolment.
Patients have been diagnosed with OAB based on the OABSS and treated with one of the following antimuscarinics for at least 4 weeks.
Propiverine hydrochloride 20 mg (orally once per day).
Imidafenacin 0.2 mg (orally two times per day).
Solifenacin succinate 5 mg (orally once per day).
Fesoterodine fumarate 4 mg (orally once per day).
Oxybutynin hydrochloride transdermal 73.5 mg (once per day, applied to the lower leg, lumbar region or thigh).
Patients are willing and able to accurately complete the voiding diary/questionnaire, including measuring urine output over a 3-day period.
Patients are willing and able to follow and comply with the study protocol, including study visits and tests.
Patients fully understand the study content and have given their written consent.
The following exclusion criteria will be used for this trial:
Patients have clinically significant bladder outlet obstruction.
Patients have high residual urine volume (>150 mL).
Patients have significant stress incontinence or mixed incontinence in which stress incontinence is the predominant factor.
Patients use a continuously placed urinary catheter or clean intermittent catheterisation.
Patients are receiving non-pharmacological treatment for urinary incontinence, including sacral nerve stimulation. However, bladder training programmes or pelvic floor muscle exercises initiated more than 30 days prior to enrolment are acceptable.
Patients are determined by the principal investigator or co-investigator to have a history of disease or surgery that affects the assessment of OAB-related voiding (eg, patients treated with intravesical botulinum toxin injection).
Patients have chronic inflammatory disease or malignant disease in the pelvic region.
Patients have undergone intravesical treatment for bladder malignancy within 12 months or have a history of bladder, prostate or uterine cancer within 5 years prior to enrolment. However, patients with a history of these cancers may be enrolled if they have been treated, are cancer-free, and have not had a recurrence in 5 years.
Patients with uncontrolled narrow-angle glaucoma, urinary retention, pyloric stenosis, severe ulcerative colitis, toxic megacolon, myasthenia gravis or any other contraindication to antimuscarinics as determined by the principal investigator or coinvestigator.
Patients have a history of hypersensitivity to the components of vibegron.
Patients are pregnant, potentially pregnant or lactating.
Patients are deemed ineligible by the principal investigator or co-investigator for medical, psychological or other reasons.
Study population and recruitment
In accordance with the inclusion and exclusion criteria, we will enrol patients with OAB who have responded insufficiently to initial 4-week antimuscarinics. The physician in charge will obtain informed consent and written consent forms from patients prior to study enrolment. The consent forms and explanatory documents provided to the participants are shown in online supplemental Additiona file 2 and online supplemental Additiona file 3.
Supplemental material
Supplemental material
After inclusion, patients will be assigned randomly to either the add-on group (vibegron added to current antimuscarinic) or the switch group (switch to vibegron from current antimuscarinic) in a 1:1 ratio. This randomisation will be based on the minimisation method, using age (≥75 years/<75 years), sex (male/female) and mean daily urinary frequency (≥10 times/<10 times) as stratification factors. The process will be conducted in an open-label fashion, using a computer-generated minimised randomisation allocation sequence as part of the electronic data capture (EDC) system.
Interventions
Figure 1 outlines the study procedures. Both groups will receive 50 mg of vibegron daily for OAB treatment. The add-on group will continue their current antimuscarinic alongside vibegron, while the switch group will discontinue their antimuscarinic and take only vibegron. This study does not use a placebo.

Design of the ADd-on or switch to Vibegron in patients with overactive bladder (OAB) InSufficiently Responding to initial 4-week antimuscarinics (ADVISR) trial.
If a patient experiences an adverse reaction to vibegron administration and/or antimuscarinics administration, its severity will be assessed using the Common Terminology Criteria for Adverse Events (CTCAE) V.5.0.15 The medication may be temporarily withdrawn or permanently discontinued based on this assessment. For grade 3 or higher constipation, dry mouth or urinary tract infection, vibegron administration will be paused until the condition improves to grade 2 or lower. Treatment can then resume. However, if these adverse events persist for more than 14 days, vibegron will be discontinued.
Follow-up, data collection and data protection
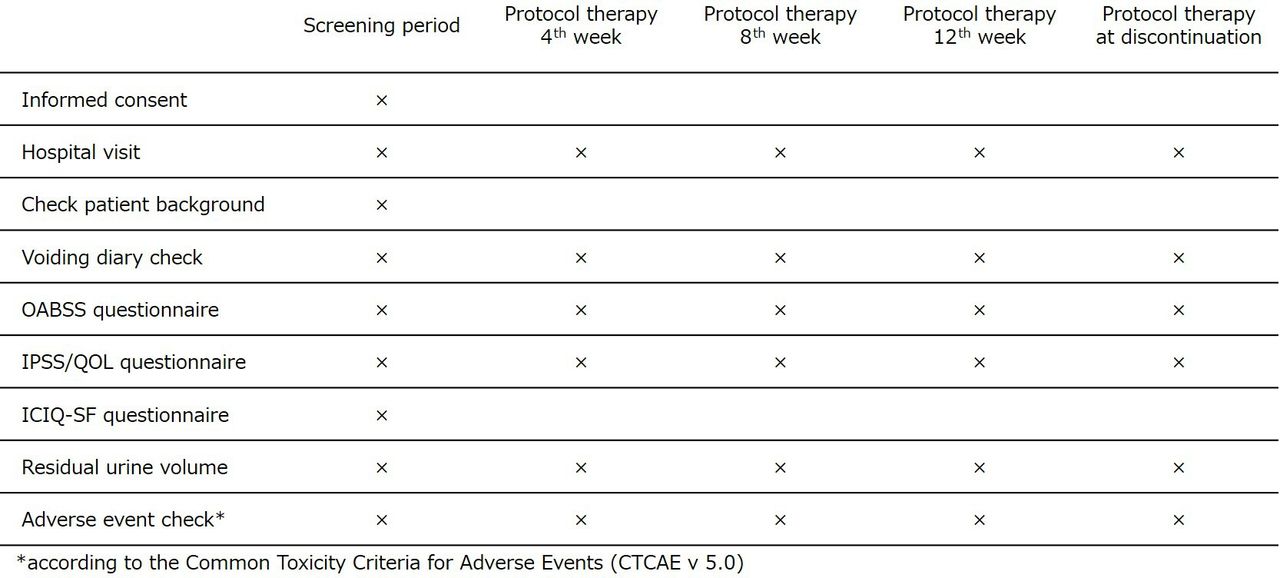
Patients will be seen every 4 weeks for 3 months following trial enrolment. At each visit, a 3-day voiding diary, OABSS, International Prostate Symptom Score (IPSS) and residual urine volume will be evaluated (figure 2). Additionally, adverse events related to treatment will be assessed using CTCAE V.5.0. To enhance monitoring adherence, the physician in charge will explain the importance of follow-up tests during patient visits and encourage participation in the study’s follow-up process. To ensure data quality, a clinical research coordinator will enter data from medical records, and central monitoring will be implemented. To safeguard patients’ personal information, each patient will be assigned a unique identification code. All data will be stored as password-protected electronic files on the EDC system before, during and after the study, with access restricted to investigators only.

{kind=link}
{kind=link}
Intervention and assessment schedule for the ADd-on or switch to Vibegron in patients with OAB InSufficiently Responding to initial 4-week antimuscarinics (ADVISR) trial according to the Standard Protocol Items: Recommendations for Interventional Trials (SPIRIT). Patients will visit the hospital every 4 weeks until approximately 12 weeks to collect data. ICIQ-SF, International Consultation on Incontinence Questionnaire-Short Form; IPSS/QOL, International Prostate Symptom Score/quality of life; OAB, overactive bladder; OABSS, Overactive Bladder Symptom Score.
Outcome measures
Primary outcome measure
The primary endpoint is the intergroup comparison of the change in daily voiding frequency between the add-on and switch groups at 12 weeks after initiating protocol treatment. Daily urinary frequency will be calculated as the average from 3-day urinary diaries immediately preceding the assessment date.
Secondary outcome measure
Secondary outcomes include:
Intergroup comparison of the change in daily urinary frequency between the add-on and switch groups at 4 and 8 weeks after protocol treatment initiation.
Intergroup comparison of the change in daytime urinary frequency between the add-on and switch groups at 4, 8 and 12 weeks after protocol treatment initiation.
Intergroup comparison of the change in nocturnal urinary frequency between the add-on and switch groups at 4, 8 and 12 weeks after protocol treatment initiation.
Intergroup comparison of the change in daily urinary incontinence frequency between the add-on and switch groups at 4, 8 and 12 weeks after protocol treatment initiation.
Intergroup comparison of the change in OABSS scores and total score between the add-on and switch groups at 4, 8 and 12 weeks after protocol treatment initiation.
Intergroup comparison of the changes in IPSS/QOL scores and total scores between the add-on and switch groups at 4, 8 and 12 weeks after protocol treatment initiation.
Intragroup comparison of the change in daily urinary frequency in both the add-on and switch groups at 4, 8 and 12 weeks after protocol treatment initiation.
Intragroup comparison of the change in daytime urinary frequency in both the add-on and switch groups at 4, 8 and 12 weeks after protocol treatment initiation.
Intragroup comparison of the change in nocturnal urinary frequency in both the add-on and switch groups at 4, 8 and 12 weeks after protocol treatment initiation.
Intragroup comparison of the change in daily urinary incontinence frequency in both the add-on and switch groups at 4, 8 and 12 weeks after protocol treatment initiation.
Intragroup comparison of the change in OABSS scores and total score in both the add-on and switch groups at 4, 8 and 12 weeks after protocol treatment initiation.
Intragroup comparison of the change in IPSS/QOL scores and total score in both the add-on and switch groups at 4, 8 and 12 weeks after protocol treatment initiation.
Safety endpoints
The safety endpoints for this study are as follows:
Research drug
The investigational drug is Beova, with the identification code 2590017F1025 and the chemical name vibegron. This study is covered by clinical research insurance. Compensation may be provided for adverse effects caused by vibegron administration that are not listed among its known possible adverse effects.
Determination of sample size
The target sample size is calculated to confirm the non-inferiority of the switch group compared with the add-on group for the primary endpoint, which is the change in daily voiding frequency at 12 weeks after initiation of protocol treatment. A Bayesian mixed effects model for repeated measures (MMRM) is employed to evaluate non-inferiority, with the success criteria defined as a posterior probability of at least 80% that the between-group difference in change in daily voiding frequency (switch group−add-on group) at 12 weeks would be less than the non-inferiority margin of one-time. With reference to the clinical trial results of mirabegron, a β3 receptor agonist,9 17 we assume that the change in daily voiding frequency at 4, 8 and 12 weeks would be −0.6,–1.25 and −1.9 times in the switch group, and −0.7,–1.4 and −2.1 times in the add-on group, respectively. Additionally, we assume a common SD of 2.1 and a correlation coefficient of 0.8. Under these assumptions, the required sample size to achieve an 80% or higher probability of meeting the non-inferiority criterion is 50 patients per group. To account for a 10% dropout rate, the target sample size is set at 55 patients per group (110 patients in total).
Statistical analysis
All analyses for efficacy endpoints will be performed in the full analysis set (FAS), with the per-protocol set (PPS) as a reference analysis population. The FAS will include all randomised patients excluding: (1) patients who have never received any protocol treatment, (2) patients who do not have any postrandomisation data and (3) patients who violate the well-defined and objectively determinable selection and exclusion criteria. The PPS will consist of the population from which the following patients are excluded from the FAS: (1) patients who were unable to properly complete the voiding diary, (2) patients who were retrospectively found to meet any of the exclusion criteria and (3) patients found to have serious protocol violations. The analysis methods for each efficacy endpoint are as follows.
Analysis of the primary outcome
The primary analysis will employ a Bayesian MMRM including the treatment group, the interaction between treatment group and time point, and the interaction between baseline value and time point as fixed effects. The covariance structure for repeated measurements within each patient will initially be specified as unstructured; however, if convergence issues arise, it will be adjusted to a first-order autoregressive or compound symmetry structure in that order. Non-informative priors will be used for all parameters, and no imputation will be applied for missing data.
Using this model, the maximum a posteriori estimator (MAP) and 95% highest posterior density (HPD) intervals for changes in daily voiding frequency at 4, 8 and 12 weeks will be derived for each group. Additionally, the MAP and 95% HPD intervals for the differences in changes between the treatment groups at these time points will be estimated. The posterior distribution of the difference in the change in daily voiding frequency at 12 weeks will be evaluated to determine whether the non-inferiority criterion is met.
Analysis of the secondary outcomes
For the secondary outcomes (1–6), the same analysis will be conducted using Bayesian MMRM as for the primary outcome. However, non-inferiority will be evaluated only for the secondary outcome (1), and the posterior probability of the difference in the change in daily voiding frequency between the treatment groups (switch group−add-on group) at 4 and 8 weeks is lower than the non-inferiority margin of one-time will also be evaluated.
For the secondary outcomes (7–12), a frequency-based MMRM will be performed. The model includes the treatment group, the interaction between the treatment group and time point, and the interaction between baseline value and time point as fixed effects. The covariance structure for repeated measurements within each patient will initially be specified as unstructured; however, if convergence issues arise, it will be modified to a first-order autoregressive or compound symmetry structure in that order. No imputation will be performed for missing data.
Using this model, the least-squares mean, 95% CI and p value for outcomes at weeks 4, 8 and 12 will be calculated for each group.
Analysis of the safety outcomes
The safety analysis will be performed in the safety analysis set (SAS) which is defined as the population that will be randomised and will receive the protocol treatment. The data from the SAS will be analysed by the actual treatment that the subject received. Adverse event data will be listed individually and summarised by the number and proportion of patients for each treatment group in the SAS. The number and proportion of patients discontinuing treatment due to adverse events are summarised in the SAS.
No interim analysis will be performed in this study.
Patient and public involvement
Patients or the public will not be involved in the design or conduct or reporting or dissemination plans of our research.
Dissemination
The study results will be submitted for publication in a peer-reviewed journal and presented at national and international scientific conferences.
Discussion
This study presents the protocol for an open-label randomised trial comparing vibegron with or without antimuscarinics in patients with OAB who have responded insufficiently to initial treatment. To our knowledge, this randomised trial is the first to assess the non-inferiority of switching to vibegron following antimuscarinic treatment in patients with OAB. The study’s significance lies in its scientific evaluation of secondary treatment options’ effectiveness for OAB.
To evaluate OAB symptoms, OABSS and IPSS were used in this study in addition to the 3-day voiding diary; IPSS has been used to assess lower urinary tract symptoms and severity of benign prostatic obstruction in men, and it is also useful to assess lower urinary tract dysfunction in women.18 Hsiao et al reported significantly higher IPSS storage subscore values found in OAB patients with UUI, and it was well correlated with OABSS in women.19
The efficacy of vibegron monotherapy in patients with OAB is well documented, with Phase III comparative studies demonstrating a significant reduction in mean daily urinary frequency compared with placebo (imidafenacin 0.2 mg/day).12 However, research on combination therapy with antimuscarinics is scarce, highlighting the need for further investigation in this area. While mirabegron, another β3 receptor agonist, has shown promising effects, studies on vibegron as a secondary treatment for OAB patients already using antimuscarinics remain limited.13
In this study, we aim to evaluate the efficacy and safety of two treatment approaches: adding vibegron to antimuscarinics and switching from antimuscarinics to vibegron. By comparing these two groups, we can determine which treatment option offers the best balance of benefits and risks. If we can demonstrate that vibegron is non-inferior as a secondary treatment for OAB, it could help address the growing issue of polypharmacy in older people. This, in turn, may significantly improve patients’ QOL and reduce healthcare costs. As an open-label randomised controlled trial with unblinded participants and therapists, this study has an inherent risk of selection bias. This limitation requires careful consideration when interpreting the results.
The frequentist approach, which evaluates treatment effects using observed data and quantifies certainty through CIs and p values, is a standard method for evaluating efficacy and safety. The ADVISR trial, unlike conventional OAB clinical trials, adopts Bayesian statistical analysis. Bayesian methods offer a valuable option for quantitatively assessing the certainty of results through the posterior distribution of the treatment effect, especially in small-scale clinical trials where hypothesis testing based on frequentist methods may be less interpretable. However, caution is needed in interpreting Bayesian results due to potential subjectivity in setting prior probabilities. The ADVISR trial addresses this using non-informative prior distributions, determining the posterior distribution primarily based on observed data. This minimises arbitrariness in prior distribution selection while enabling efficacy evaluation. The adoption of Bayesian statistics in the ADVISR trial is expected to provide deeper insights into OAB treatment and more precise treatment effect evaluation. This innovative approach could transform the entire process from clinical research design to result interpretation and clinical decision-making, potentially setting a new standard for more patient-centred, effective and efficient clinical trial designs. The results of this study are expected to provide a solid foundation for future large-scale prospective randomised trials.
Supplemental material
Ethics statements
Patient consent for publication
Footnotes
Contributors All authors contributed to the conception and conduct of this clinical trial. TY and SY designed and wrote the protocol and the manuscript. MI and HS designed and discussed the protocol. YM, TT, MT, HF, SU, SA, MI, SK, AY, NK, RT, YS, YO, TT, KN, HT, RKo and YF helped with implementation. MI will monitor the data set. RH and RKi designed the statistical analysis. SY is the principal investigator. TY is responsible for the overall content as guarantor. All authors contributed to and approved the final version of the manuscript.
Funding This work is supported by KYORIN Pharmaceutical Co, (Tokyo, Japan) and KISSEI Pharmaceutical Co, (Nagano, Japan). KYORIN Pharmaceutical Co, and KISSEI Pharmaceutical Co, had no input on the design of the study, collection of data, analysis, interpretation of data or in writing the manuscript.
Competing interests None declared.
Patient and public involvement Patients and/or the public were not involved in the design or conduct or reporting or dissemination plans of this research.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.