Article Text

Abstract
Introduction Therapeutic interventions for disorders of consciousness lack consistency; evidence supports non-invasive brain stimulation, but few studies assess neuromodulation in acute-to-subacute brain-injured patients. This study aims to validate the feasibility and assess the effect of a multi-session transcranial alternating current stimulation (tACS) intervention in subacute brain-injured patients on recovery of consciousness, related brain oscillations and brain network dynamics.
Methods and analyses The study is comprised of two phases: a validation phase (n=12) and a randomised controlled trial (n=138). Both phases will be conducted in medically stable brain-injured adult patients (traumatic brain injury and hypoxic-ischaemic encephalopathy), with a Glasgow Coma Scale score ≤12 after continuous sedation withdrawal. Recruitment will occur at the intensive care unit of a Level 1 Trauma Centre in Montreal, Quebec, Canada. The intervention includes a 20 min 10 Hz tACS at 1 mA intensity or a sham session over parieto-occipital cortical sites, repeated over five consecutive days. The current’s frequency targets alpha brain oscillations (8–13 Hz), known to be associated with consciousness. Resting-state electroencephalogram (EEG) will be recorded four times daily for five consecutive days: pre and post-intervention, at 60 and 120 min post-tACS. Two additional recordings will be included: 24 hours and 1-week post-protocol. Multimodal measures (blood samples, pupillometry, behavioural consciousness assessments (Coma Recovery Scale-revised), actigraphy measures) will be acquired from baseline up to 1 week after the stimulation. EEG signal analysis will focus on the alpha bandwidth (8–13 Hz) using spectral and functional network analyses. Phone assessments at 3, 6 and 12 months post-tACS, will measure long-term functional recovery, quality of life and caregivers’ burden.
Ethics and dissemination Ethical approval for this study has been granted by the Research Ethics Board of the CIUSSS du Nord-de-l’Île-de-Montréal (Project ID 2021–2279). The findings of this two-phase study will be submitted for publication in a peer-reviewed academic journal and submitted for presentation at conferences. The trial’s results will be published on a public trial registry database (ClinicalTrials.gov).
Trial registration number NCT05833568.
- Adult intensive & critical care
- Neurology
- Adult neurology
- Neurological injury
- Neurophysiology
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Strengths and limitations of this study
This study assesses the feasibility and preliminary effectiveness of a non-invasive therapeutic intervention to promote recovery and prevent prolonged disorders of consciousness in acute-to-subacute brain-injured patients.
The experimental design uses multimodal short-term tracking to capture changes in consciousness recovery status.
Longitudinal assessments are performed at 3, 6 and 12 months to assess functional outcomes and quality of life of participants, as well as the perceived burden of their caregivers.
The study is limited by the high variability of brain lesions between aetiologies, as differential tissue conductivity affects the injected current’s trajectory within the brain.
Introduction
After a severe acquired brain injury (ABI) such as severe traumatic brain injury (sTBI), stroke or global hypoxic-ischaemic encephalopathy (HIE) following post-cardiac arrest, surviving patients may either fully emerge from a coma, transition to a disorder of consciousness (DoC) characterised by varying degrees of impairments in arousal and/or awareness, or ultimately experience brain death.1–4 The first 28 days post-injury, known as acute care,5 present significant challenges as crucial therapeutic decisions are made and end-of-life considerations often arise. Predicting outcomes during this period is notoriously difficult because of the diverse phenotypes within the population and high variability in injuries,6 7 and the limited usefulness of various clinical tools available that are unable to detect covert awareness.1 The temporal progression of patients with DoC involves transitioning from the acute stage after the injury to the subacute stage, where gradual improvements in arousal and awareness may occur and then to the chronic stage, which involves long-term management and potential functional recovery.1 5 More recently, Cognitive Motor Disorder has been described for patients with preserved awareness that can only be detected using non-behavioural measures of brain activity such as functional MRI or electroencephalography (EEG).6 8 9
Neuroimaging of brain activity has provided valuable insights into the underlying mechanisms of DoC, expanding possibilities for identifying targets for therapeutic intervention. Studies have revealed that coma is often caused by a disruption of neurotransmission to neocortical and thalamic neurons, which results in a decrease in cortical excitatory synaptic activity.1 10 11 This leads to disfacilitation, a phenomenon characterised by a reduction in excitatory synaptic activity that downregulates neuronal firing rates.10 12 13 Restoration of the corticothalamic system’s circuitry is considered as potentially key to behavioural emergence from a DoC.1 14–16 The anterior forebrain mesocircuit/default mode network and the frontoparietal network have been explored for their association with cognitive recovery.15 17–20 These networks are highly interconnected, and as patients transition from lower levels of consciousness toward cognitive recovery, an associated augmentation in metabolic efficiency and strengthening of functional connectivity has been observed.15 21–23
Patients with DoC have reduced-to-absent alpha (8–13 Hz) oscillations compared with healthy individuals.24–31 Alpha activity is dominant in the parieto-occipital cortex during periods of wakeful rest in healthy people, and it is believed to originate from thalamocortical circuits.32–39 Damage to these circuits and related networks may explain this reduced alpha activity.1 28 40 In instances of thalamic and cortical deafferentation, alterations in the EEG power spectrum are observed, particularly in the alpha bandwidth, reflecting the changes in neuronal firing patterns that correspond to the severity of deafferentation.1 24 41 In patients with DoC following severe brain injury, resting-state EEG analyses have shown an important reduction in alpha power at the expense of an increase in slow-wave activity. This shift in brain activity intensifies with the severity of brain injuries (<1 Hz for more severe cases such as unresponsive wakefulness syndrome (UWS)), which likely reflects the degree of cortical deafferentation.24 27 28 The ‘ABCD’ model is based on this framework to categorise the severity of DoC according to the changes in the EEG power spectrum, but also to predict the potential for recovery.1 41 However, the disruption of dynamics in the alpha bandwidth following cortical injuries extends beyond spectral properties, as evidenced by functional connectivity analyses.27 42 43 Indeed, alpha-band networks constructed on phase-based functional connectivity, specifically weighted Phase Lag Index (wPLI), have shown a lack of integrity and robust connectivity in patients with DoC compared with healthy brains.27 43 The modulation of alpha brain oscillations is an active area of research because of its potential involvement in internal cognitive processes, including attention, perception, memory retention and awareness.34 44–50
Transcranial electrical current stimulation (tES) involves the application of a low-intensity electrical current through the scalp using two or more electrodes and includes several neuromodulation techniques.33 34 Anodal transcranial direct current stimulation (tDCS) has been used in patients with DoC, and while most study outcomes have been estimated using behavioural scales, recent studies have explored its electrophysiological effects.1 51–56 tDCS has the potential to induce polarity-specific modulation in neuronal excitability if sustained for several minutes, resulting in long-term potentiation attributed to synaptic plasticity.57 58 Other forms of tES, such as high-frequency transcranial random noise stimulation (tRNS) and transcranial pulsed-current stimulation (tPCS), have been explored as potential therapeutic interventions to promote recovery of consciousness in patients with DoC.59–61 tRNS comprises applying a current that varies randomly in frequency and amplitude, while tPCS corresponds to electrical stimulation as pulses.61 62 Both studies did not result in significant behavioural or electrophysiological improvements compared with the sham condition in patients with DoC59 60 In contrast, transcranial alternating current stimulation (tACS) is characterised by the injection of a sinusoidal current through the scalp.63–65 Two main hypotheses have been suggested to explain the mechanisms underlying tACS. First, the entrainment effect, which suggests that administration of the sinusoidal current leads to a synchronisation with the endogenous brain activity. Second, the long-lasting effects of tACS would result from neuroplastic changes, occurring in response to the alternating polarisation of the membrane potential of neurons.32 33 66 67 Its ability to modulate brain activity offers promising versatility, as it can target specific brain oscillations implicated in cognitive and motor functions.32 68–72 tACS has shown promise in selectively increasing the amplitude of alpha oscillations in healthy individuals.32 Replicating this effect in individuals with DoC in the acute-to-subacute phase may be a promising therapeutic approach to recovery of consciousness and cognitive function.
Interventions targeting the acute-to-subacute phase of DoC may be critical for functional recovery. Non-invasive brain stimulation techniques have shown promising results, though most study results are limited to the chronic stages of the condition.1 51 Developing effective interventions during the acute phase, a time-window prone to neuronal plasticity, may improve long-term functional outcomes and enhance the quality of life for patients and their families. While previous research has reported improvements in the Coma Recovery Scale revised (CRS-R) with the use of non-invasive electrophysiological interventions in prolonged DoC patients,51–53 it remains necessary to assess if these interventions can positively impact outcomes when applied in the acute stage.
Our study aims to achieve two main goals via two phases. In the validation phase, we first aim to assess the feasibility of a multi-session tACS intervention in acute-to-subacute DoC individuals in the intensive care unit (ICU). In the second phase we aim to conduct a double-blind randomised controlled clinical trial based on the validation phase’s results, to assess the efficacy of a multi-session tACS intervention on recovery of consciousness, related brain oscillations and network dynamics in subacute patients with brain injuries. As a tertiary objective, this study aims to evaluate the effectiveness of the intervention protocol through repeated assessment of complementary measures of different modalities. Repeatedly measuring behavioural consciousness, pupil dilatation and the rest-activity cycle will provide complementary monitoring and tracking of consciousness recovery. As a fourth objective, this study aims to evaluate the effectiveness of the intervention protocol on long-term functional recovery outcomes. This study has the potential to provide a new therapeutic approach proactively seeking to prevent the evolution of acute DoC into a prolonged condition.
Methods
Ethics and study design
This study was approved by the Research Ethics Board of the CIUSSS du Nord-de-l’Île-de-Montréal (Project ID 2021–2279). The study will be conducted in two phases: a prospective observational validation phase (Vanguard study) and a parallel group double-blind randomised controlled trial. As the participants are unable to provide informed consent due to their medical condition, their legal representatives will be informed about the study and will provide consent following the principles of the Declaration of Helsinki. Should the participant demonstrate the capacity to provide informed consent while participating in the protocol participation, their consent will be required to continue participation in this study (an example of the patient consent form is provided).
Patient involvement
The objective of the validation phase (Vanguard study) is to allow participants’ caregivers to give input about the study design, its management and materials to improve the protocol’s feasibility. In case participants recover awareness during the study, we will ensure that informed consent is obtained to pursue and provide updates as well as maintain open communication with them and their caregivers to prioritise participants’ well-being throughout the study.
Participants
For the Vanguard study, 12 (n=12) medically stable brain-injured, non-sedated patients will be recruited from the ICU of the Hôpital Sacré-Coeur de Montréal, a university-affiliated Level 1 Trauma Centre in Montreal, Quebec, Canada. For the clinical trial phase, the estimated sample size is 138 (n=69 per group) based on a Student’s t-test derived from a randomised, sham-controlled clinical trial comparing a 4-week transcranial direct current stimulation protocol on patients with DoC.73
Inclusion criteria
To be eligible for the study, participants must:
Be adults aged 18 or older.
Have a Glasgow Coma Scale (GCS) score remaining ≤12 at least 24 hours after the withdrawal of continuous sedation (if applicable). Once participants are screened and identified as potentially eligible, a CRS-R assessment will be conducted to confirm a DoC.
Have a severe ABI of traumatic or non-traumatic aetiology (ie, sTBI, HIE or subarachnoid haemorrhage) and treated at the ICU or intermediate ICU.
Exclusion criteria
Patients will be excluded from the study if they have:
Severe medical comorbidities/complications, such as stroke and status epilepticus.
Focal brain lesion(s) in the occipital and parietal lobes (ie, the stimulation site), even if unilateral based on accessible clinical brain imaging reports.
Pre-existing severe neurological conditions/disorders involving cognitive deficits such as neurodegenerative diseases (eg, Amyotrophic Lateral Sclerosis, dementia, Parkinson’s), hereditary conditions (eg, Huntington’s Chorea), central nervous system disorders.
Constant and intense agitation preventing proper application of the tACS and EEG equipment.
Invasive neurological monitoring (intracranial pressure and/or brain tissue oxygenation).
History of epilepsy (patient with a seizure episode in response to non-exclusive active tACS intervention).
Aneurysm clip(s).
Subdural brain electrodes.
Metallic brain implant.
Implantable neurostimulator.
Craniectomy with no bone flap replacement.
Cervical collar limiting access to the occipital brain region.
Participation in a current (or previous) study that may have a confounding effect, as assessed by the research team.
Screening, recruitment and activation
To facilitate recruitment and obtain consent, the research team will approach family members of patients who meet the inclusion criteria before meeting the protocol activation criteria: (1) a GCS≤12 (ie, persistent impaired awareness), 24 hours after cessation of continuous sedation and (2) patient’s clinical stability confirmed by the medical team. The activation of the study for patients who have received sedatives (eg, propofol, benzodiazepine (midazolam), ketamine), sodium or calcium channel blockers (eg, carbamazepine), or N-méthyl-D-aspartate receptor antagonists (eg, dextromethorphan) will depend on specific time windows according to the type of molecule administered. The following procedure was determined by a trained ICU pharmacist, ensuring specific time windows are adhered to, to limit interactions between tACS and medication: If continuous sedation with benzodiazepines has been administered:
Less than 48 hours: 2 days before activating the protocol.
Between 48 and 96 hours: 3 days before activating the protocol.
96 hours or more, or in the presence of severe renal insufficiency (creatinine clearance <30 mL/min): 5 days before activating the protocol.
In the case of a single administration during the protocol, the following time intervals should be followed before administering the protocol:
Propofol: 3 hours.
Midazolam: 12 hours.
Dexmedetomidine: 2 hours.
Ketamine: 6 hours.
Randomisation and allocation
In this parallel-group randomised controlled trial (RCT), participants will be allocated using computer-generated random numbers through a centralised web-based system. Two groups, Group A (active tACS) and Group B (sham), will have an equal 1:1 ratio and stratification based on aetiology, sex and age will ensure balanced representation. The allocation sequence will be implemented using sealed envelopes to conceal assignment by a trained staff and an independent committee will monitor the process for protocol adherence and data quality. To ensure blinding of the outcome assessor to condition allocation, intervention protocols will be pre-programmed to only be labelled as A or B on the computer screen and will have the same procedure for implementation. Analyses will be done blinded to the condition allocation, by only using group labels A and B. On request, participants will be informed of their group assignment after their 12-month follow-up assessment.
Protocol
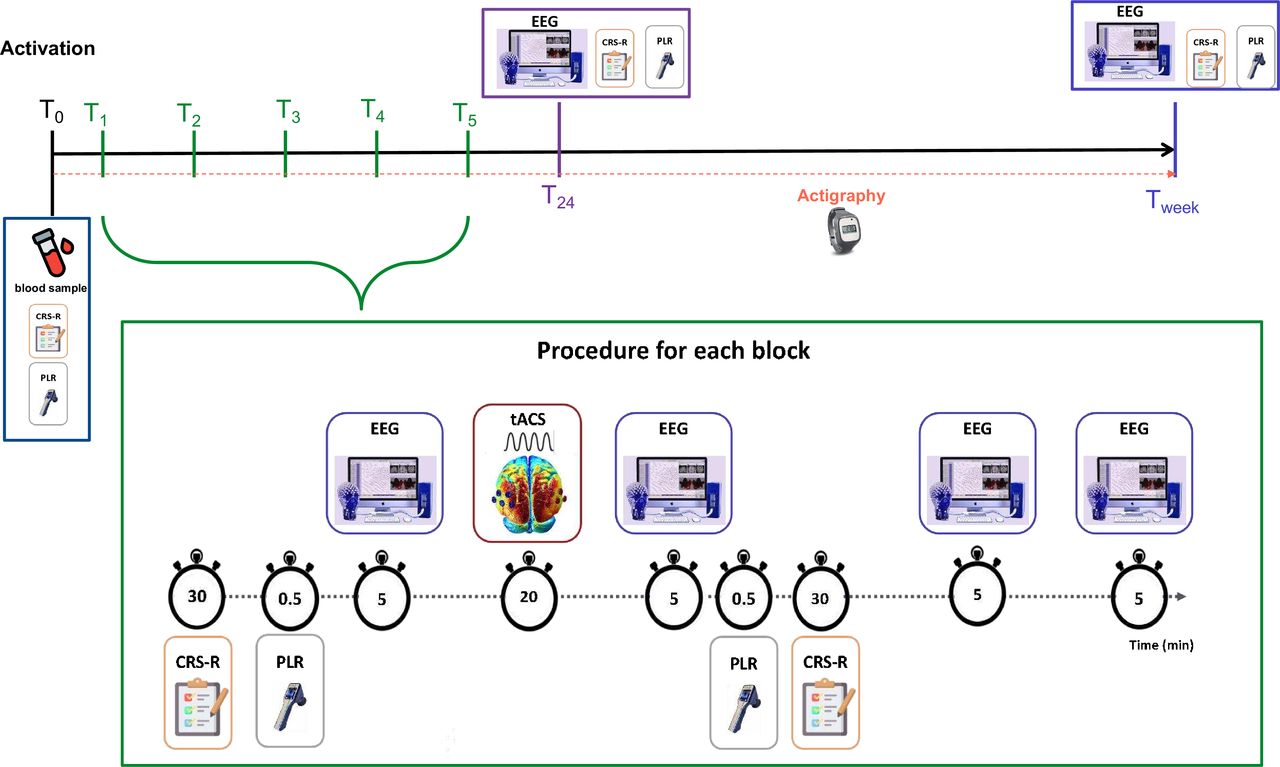
See figure 1 for an overview of the timeline for data collection and assessments. On activation, baseline measures (T0) including blood sample, actigraphy installation, initial CRS-R and pupillometry will be acquired. The intervention and data collection, including the high-density EEG recordings and the 20 min tACS/sham block detailed in figure 2, will be repeated for 5 days, starting from T1 and ending on T5. The 5 min resting-state EEG recordings will be conducted as follows for the five daily tACS’ sessions: (1) baseline recording (prior to tACS) (pre0) (2) post-tACS recording (directly after tACS administration) (post0); (3) at 60 min after the tACS’ session (post60); and (4) at 120 min after the tACS’ session (post120). At both 24 hours and 1 week after the fifth tACS’ session (Tweek), 5 min resting-state EEG will be recorded (T24) along with measures of CRS-R and pupillometry. To promote participant retention, family members will be allowed to observe daily sessions and will be in constant communication with the research team will be maintained to address concerns and provide updates. Following this procedure, we will prospectively track the functional outcome, the burden of care for caregivers and the health-related quality of life (EuroQol 5-Dimension 5-Level (EQ-5D-5L)) at 3, 6 and 12 months after the intervention protocol, via phone interviews with participants and caregivers or next of kin.

Overview of the protocol. Overview of the timeline of study measures. Brain-injured patients who remain at a Glasgow Coma Scale score of ≤12 after the withdrawal of continuous sedation (if applicable) will be recruited from the ICU. The experimental protocol will include several measures at each time point. Brain activity will be recorded before and after each intervention block (20 min transcranial alternating current stimulation/sham), using high-density EEG. A phone assessment will occur at 3, 6 and 12 months post-participation to track long-term outcomes using the Glasgow Outcome Scale-Extended (severe traumatic brain injury)/cerebral performance category (hypoxic-ischaemic encephalopathy following cardiac arrest), the Disability Rating Scale (DRS) and the Functional Independence Measure (FIM); EuroQol 5-Dimension 5-Level (EQ-5D-5L); the Burden Scale of Family Caregivers (BSFC). CPC, cerebral performance category; CRS-R, Coma Recovery Scale revised; EEG, electroencephalography; GOS-E, Glasgow Outcome Scale-Extended; ICU, intensive care unit.

{kind=link}
{kind=link}
Timeline for repeated intervention sessions and procedure for each block. Overview of the full intervention protocol. T0 corresponds to the baseline measures and actigraphy installation. Days corresponding to T1 to T5 include the detailed procedure for the tACS/sham protocol with specific duration estimations. Before and after each intervention block, a CRS-R assessment, pupillometry measures and 5 min resting-state EEG will be acquired. To measure potential aftereffects of the intervention, an additional 5 min resting-state EEG recording will be conducted at 60 and 120 min after the 20 min intervention. T24 consists of the acquisition of measures (ie, of CRS-R EEG and pupillometry) 24 hours after the last stimulation condition session and Tweek is after a week. CRS-R, Coma Recovery Scale revised; EEG, electroencephalography; PLR, pupillary light reflex; tACS, transcranial alternating current stimulation.
Demographic and clinical characteristics
To characterise each group, the following information will be documented: age, sex, occupation, level of education, ethnicity, languages spoken, pre-morbid medical history (including neurocognitive and psychiatric disorders), pre-morbid substance use, aetiology type (eg, sTBI or HIE), types of brain injury, mechanism of injury, GCS at initial admission, CT scan and/or MRI findings (eg, Marshall and Rotterdam scores); sedative molecules administered (ie, name of molecules and cumulative dosage received since admission), analgesia drug levels (name and dosage); delirium duration; post-traumatic amnesia duration; ICU stay duration; and hospital stay duration. We expect a significant heterogeneity in brain lesions consequently to sTBI, as sTBI encompasses various brain injuries with varying localisations. This includes—although is not limited to—diffuse axonal injury, haematomas (subdural, subarachnoid, epidural), haemorrhages, contusions and oedema. Additionally, these injuries can lead to direct medical complications such as strokes, vascular dissections and pulmonary embolisms. These specific brain injuries and their consequences are not dissociable and will all be thoroughly reported in our study. In contrast, anoxia resulting from post-cardiac arrest primarily leads to an HIE brain injury. In addition to the baseline pharmacological dosage at T0, all doses, types, administration routes and times for all sedatives and analgesics received in the 48 hours prior to and up to the completion of the protocol will be documented.
Measures
See figure 2 for the sequential procedure.
Blood sample
At T0, a blood test will be conducted by the medical staff, then analysed to confirm the absence of pharmacological agents that could interfere with the intervention as we assess its efficacy.74 This will enable us to control for molecules administered at the time of EEG recordings and tACS/sham administration. Two 6 mL tubes will be used for blood collection and centrifuged after 30 min at 3000 revolutions per minute (RPM) for 15 min at 5°C, with the top layer containing plasma retained. Aliquots of 0.5 mL of plasma will be prepared in cryotubes and stored at −80°C until use.
Pupillometry
Pupillometry will be used as a non-invasive complementary measure to evaluate the effects of tACS in patients with DoC and monitor their autonomic nervous system activity. In previous studies with DoC, pupillary measurements have been found to be a relevant tool for the assessment of consciousness, as responses could be decoded from pupillary measurements following verbal commands.75 The NeuroLight (IDMED, Marseille, France) pupillometry device will collect the pupillary light reflex which involves measuring pupillary constriction in response to a light stimulus,76 before and after each stimulation session. Constriction velocity, which measures the rate at which the pupil changes from its maximum size at rest to its minimum size following the induction of a light stimulus, and constriction latency, which is the time between the onset of the light stimulus and the start of pupillary constriction, will also be measured.76
Actigraphy
Actigraphy provides a non-invasive and accessible way to assess the effects of tACS stimulation on the recovery of the rest-activity cycle, and is a proxy measure of the sleep-wake cycle.77 This device can be placed on the wrist and features an integrated multi-axis accelerometer to monitor motion. It can be worn for extended periods, even weeks. Sleep and wakefulness periods are estimated based on the frequency and intensity of movements.78 Perturbations of sleep-wake cycles are prevalent among patients with DoC, and there is a well-documented relationship between the recovery of consciousness and the restoration of sleep-wake cycles.79–83 Duclos and colleagues84 highlighted the relevance of using actigraphy devices to measure the consolidation of the 24-hour sleep-wake cycle in patients with moderate or sTBI. The recovery of the 24-hour wake-sleep cycle consolidation has been shown to coincide with improvements in patients’ state of consciousness.84 85 Either an Actiwatch-L or Actiwatch-Spectrum device (Actiwatch-L or Actiwatch-Spectrum; Philips Healthcare, Andover, Massachusetts, USA) will record additional measurements. The device will be placed on the patient’s non-dominant (non-paralysed if applicable) wrist at T0 of the stimulation protocol for baseline assessment. Patients will wear the device throughout the protocol for a measurement period of 14 days. The device records degree and intensity of movement intensity and degree through an integrated multi-directional accelerometer, while it also measures surrounding light intensity via a photodiode.86 Key variables of the sleep-wake cycle, such as total sleep time and wakefulness time, can be estimated using a software to analyse the collected data. The use of actigraphy as a proxy of the sleep-wake cycle has been validated in hospitalised trauma patients, including those with brain injuries.87
Behavioural consciousness measurements
Coma Recovery Scale-revised
The CRS-R is a standardised and validated tool for assessing patients with DoC.4 88 It identifies four categories: coma (absence of arousal and awareness), the UWS (presence of arousal without behavioural signs of consciousness), the minimally conscious state (MCS—presence of arousal and fluctuant but reproducible signs of awareness)) and the emergence from MCS (presence of functional communication or functional object use).88 MCS is divided into two subcategories based on the absence (MCS−) or presence (MCS+) of residual language function.89 90 Emergence from MCS is identified by either reliable functional communication or object use.88 The CRS-R provides a more comprehensive evaluation than the GCS with greater granularity in assessing auditory, visual, oromotor/verbal function, communication and arousal.91–93 The CRS-R is comprised of six subscales designed to measure auditory functions, receptive and expressive language, communication skills, visual perception, motor functions and arousal, for 23 items.94 The subscales are hierarchical, with low scores associated with reflexive behaviours and high scores reflecting behaviours resulting from cognitive mediation.94 These scores range from a minimum of 0 to a maximum of 23. Scoring is standardised according to criteria of the presence or absence of clearly discernible, defined functional behaviours.94 The CRS-R is sensitive enough to distinguish between MCS+ and MCS−, with MCS+ reflecting less functional impairment than MCS− in terms of language expression and reception skills.94 95
Glasgow Coma Scale
The GCS was developed initially to assess the level of consciousness objectively in patients, based on three main components: Motor response (maximum of six points), verbal response (maximum of five points) and eye opening (maximum of four points).96–98 This scale is used in the ICU as a monitoring tool. Although the CRS-R is the gold standard assessment tool for disorders of consciousness,88 the GCS scores will be assessed in parallel to the CRS-R assessments, given that it is the standard monitoring tool in the ICU.
Full Outline of UnResponsiveness
We will use the Full Outline of UnResponsiveness scale when participants are intubated and objectively unresponsive, as it has been validated to be effective in detecting the locked-in syndrome and the UWS.99 This scale comprises four subscales that assess different domains of neurological function, including eye response, motor response, brainstem reflexes and breathing patterns, with a total score of 17.99 100
High-density EEG
The EEG data will be recorded using a 128-Channel Geodesic Sensor Net (Magstim-EGI, Oregon, USA) with sponge-based electrode nets, with a reference to Cz. This type of EEG system is portable and can be set-up at the bedside in approximately 10 min. The impedance at the electrode-skin contact will be kept below 50 kΩ and monitored before each recording and tACS/sham block. Before tACS administration, resting-state brain activity will be recorded for 5 min. As the EEG system is a hybrid, the recording electrodes also act as stimulation electrodes. Between recording sessions, the EEG net will be removed to allow participants to rest for 45 min between post60 and post120.
Intervention
Transcranial alternating current stimulation
The tACS will be administered using the same setup as the EEG acquisition system (GTEN 200, Magstim-EGI, Oregon, USA). The current will be delivered through a current controller connected to an amplifier via electrodes from the EEG net. To numb any unpleasant sensation but keep conductive properties, a mix of a lidocaine gel (4%, Deep Relief) and conductive paste (Ten20) will be applied under stimulating electrodes prior to each intervention block.
The stimulation parameters of this study are based on a study that achieved modulation of alpha activity in healthy individuals.101 A bilateral electrode montage of five electrodes per hemisphere over parieto-occipital cortical sites will administer tACS. The specific stimulation electrodes for the right hemisphere will be E83, E90, E96, E84 and E91, while those for the left hemisphere will be E58, E65, E70, E66 and E59 (see figure 2 for a current modelling simulation view of this montage). The intensity of the applied alternative current will be a maximum of 1 mA peak-to-peak, and the stimulation frequency will be adjusted to 10 Hz (the median value of the alpha frequency band) for both active tACS and sham conditions. Current density modelling was calculated a priori based on the stimulation parameters (ie, electrode’s surface and configuration, and current’s intensity).32 102 In the active tACS condition, the stimulation will include a 2 min ramp-up, a 20 min administration of 10 Hz at 1 mA and a 2 min ramp-down at the end of the 20 min period. The sham condition will involve a 2 min ramp-up before the electrical current is turned off, followed by a 2 min ramp-down at the end of the 20 min period during which no current will be administered. To conceal the stimulation condition administered and preserve blinding, the sham condition consists of the same ramp-up and down duration to mimic active stimulation sensations. Of note, tingling sensations associated with active stimulation are mostly reported during stimulation ramp-up and ramp-down. tACS is considered safe if guidelines and safety protocols are administered. However, potential side effects such as skin irritation and headaches have been reported in some cases. Any adverse events or unintended effects will be monitored and documented throughout the study.
Long-term outcome measures
To track and compare the trajectory of long-term recovery between tACS and sham stimulation conditions, recovery progress will be characterised through different modalities. The long-term outcome assessment will include functional recovery, perceived health-related quality of life (EQ-5D-5L),103 104 as well as the subjective Burden Scale for Family Caregivers.105 Functional outcome measures will include the Glasgow Outcome Scale-Extended for sTBI106 107 and the cerebral performance category108 for HIE, the Disability Rating Scale109 and the Functional Independence Measure (FIM).110 In the case of withdrawal of care preventing from completing long-term assessments, all relevant factors and clinical assessments that influenced the decision will be recorded.
EEG data analysis
EEG data will be pre-processed using MNE software.111 Data will be bandpass filtered between 1 Hz and 55 Hz and a notch filter will be applied at 60 Hz. Bad channels will be removed manually on inspection and data will be segmented into 10 s epochs. Epochs with excessive levels of artefacts will be manually removed by a trained experimenter. To clean the signal from non-physiological and physiological artefacts, such as muscle and eye movements, a semi-automated independent component analysis (ICA) pipeline will be used. Data will be average-referenced and non-brain electrodes will be removed for subsequent analyses.
An automated pipeline (MNE111) will calculate spectrograms ranging from 1 Hz to 50 Hz, and topographic maps of each frequency band (delta (1–4 Hz), theta (4–8 Hz), alpha (8–13 Hz), beta (13–30 Hz)). Functional connectivity changes across time, as well as brain network hubs in the alpha frequency band (8–13 Hz)112–116 will be calculated using the wPLI117 118 and directed Phase Lag Index119 according to Duclos and et al (2020).120
Statistical analyses
Sample size estimation
For the clinical trial phase, the estimated sample size of 138 (n=69 per group) is based on a study that used repeated tDCS in chronic DoC patients.73 Prior to the clinical trial phase, this estimated sample size will be adjusted according to feasibility (ie, number of patients in this condition yearly (~20), recruitment rates and retention).
Primary outcomes
For the validation phase, the single-site recruitment rates over a 1-year period will be calculated based on the number of participants who were assessed for eligibility, the number of participants who met the inclusion criteria and the number of participants who enrolled in the study. Retention rates will be calculated based on the number of participants who completed the intervention protocol as well as the longitudinal measures. Attrition rates and withdrawal of care will also be documented.
Linear mixed-effects models (LMM) will be used for the primary outcome analyses if assumptions are met. Active 10 Hz-tACS and sham stimulation conditions will be compared based on spectral power change in the alpha bandwidth between baseline (T1) resting-state measures and at the end of the 5-day stimulation protocol (T24) and at week after (Tweek), according to different brain regions (frontal, central and parieto-occipital). Additionally, both conditions will be compared based on the changes in functional connectivity across time (T1, T5, Tweek).
Secondary outcomes
Within-day analyses using an LMM will also be conducted to measure and compare the short-term impact of the stimulation condition on spectral and functional connectivity properties within-subject across time ((pre0)-(post0); (pre0)-(post60); (pre0)-(post120)), as well as on CRS-R assessments.
Tertiary outcomes
CRS-R
An LMM will be used to compare the CRS-R modified score121 trajectories throughout the intervention protocol between conditions (10 Hz tACS and sham) at two different time points: (1) CRS-R score at T24 compared with the baseline assessment (best score on T0 and T1); and (2) CRS-R score at Tweek compared with the baseline assessment (best score on T0 and T1).
CRS-R and actigraphy
Repeated measures analyses using LMM will be performed to compare CRS-R modified scores according to each actigraphy variable (ie, daytime activity ratio,77 87 122 night-time sleep duration and night-time fragmentation index) averaged per day.
CRS-R and pupillometry
Correlation analyses will be performed to assess the association strength between the daily pupillary light reflex measures and the CRS-R modified scores across time.
Longitudinal outcome measures analyses
Functional recovery, subjective quality of life and the burden of family caregivers’ respective trajectories across time points (3, 6 and 12 months) will be compared between conditions (active 10 Hz tACS and sham) and aetiologies (sTBI and HIE) using a three-way mixed (BBW, ie, two between-subjects factors and one within-subjects factor) analysis of variance if required assumptions are met.
Discussion
This interventional study represents the first-ever evaluation of the effects of a 5-day 10 Hz tACS protocol on associated brain oscillations, network dynamics, EEG spectral power and network properties in acute-to-subacute individuals with DoC. The proposed study also seeks to evaluate short-term behavioural responses and long-term functional and cognitive recovery outcomes resulting from the intervention. Studies that successfully modulated alpha activity in healthy individuals using tACS32 121 inspired the approach of targeting the modulation of altered brain rhythms. The study’s comprehensive and rigorous repetitive measurements may provide valuable insights into the underlying pathophysiology of DoC and help optimise treatment avenues.
This study protocol has several limitations. First, there is a high heterogeneity between brain lesions, even with only two targeted aetiologies. Therefore, further analyses will be conducted by stratifying based on aetiology (ie, sTBI and HIE). This data analysis strategy will help determine the influence of the underlying mechanism of injury and lesions’ characteristics on the trajectory of current and its dispersion, as well as responsiveness to the modulation. Second, patients will also vary in age, which will affect their recovery trajectories. Intervention and control groups will be balanced and randomised based on aetiology, sex and age to address this limitation. This type of study inherently includes convenience sampling, which is a limitation to the external validity of this study’s future results. Although the city in which the recruitment will take place is quite diverse regarding ethnic backgrounds, we will mitigate potential bias to generalisability and increase transparency by clearly characterising and defining the recruited population in addition to outlining any potential generalisability limitations. We will also use a mixed-model design for the statistical analyses, analysing all physiological and behavioural measures within-subject. Third, focal brain injuries may lead to substantial hemispheric differences in the EEG analyses because of changes in tissue conductivity properties. Unilateral hemisphere analyses will therefore be conducted in addition to whole brain analyses so that only the healthier hemisphere can be analysed in such cases. Fourth, some patients may be subject to a withdrawal of life-sustaining treatment (WoLST), which would prevent completion of follow-up outcome measures at 3, 6 and 12 months post-injury. This can introduce a negative bias on the long-term outcome comparison between the conditions (sham vs active). When conducting the analyses, the cause and the context of death will be documented thoroughly to differentiate WoLST from natural death.
This longitudinal study has the potential to improve therapeutic intervention to promote recovery in subacute patients with impaired consciousness and prevent progression into a prolonged state. The modulation of brain activity using a non-invasive brain stimulation (NIBS) approach during the acute to the subacute stage is considered an optimal clinical window, as the brain undergoes high plasticity. Since tACS is non-invasive and presents few side effects, it constitutes a potential therapeutic avenue for the treatment or prevention of prolonged DoC, as there is no standardised treatment available for this condition. The development of effective non-invasive interventions that can promote restoration of consciousness and functional abilities, even partially, can significantly improve the quality of life of patients and caregivers. Finally, tES techniques are inexpensive in comparison with other types of NIBS, especially if they diminish the prevalence of long-term care in this population, which might reduce the financial burden for both the healthcare system and the families.
Data management and access statement
To ensure data quality, a double data entry process will be implemented for all measurements. The collected data will be stored on an on-site secured network infrastructure with regular backups on a maintained, dedicated server. To ensure confidentiality, each participant will be assigned a unique anonymised ID, and the corresponding file linking to their original information will be encrypted and securely stored. For the RCT phase, a Data Monitoring Committee will be formed of clinical experts to ensure safety and provide clinical insight and an independent statistician to analyse and interpret data objectively. Independently from the investigators, an auditing procedure will be performed yearly after the initiation of the RCT.
Ethics and dissemination
Ethical approval for this study has been given by the Research Ethics Board of the CIUSSS du Nord-de-l’Île-de-Montréal (Project ID 2021–2279).
This study will be conducted at a university-affiliated Level 1 Integrated Trauma Centre, renowned for advancing clinical research with a focus on enhancing critical care practices. To disseminate the findings, the study’s results will be shared through submissions to peer-reviewed academic journals and presentations at relevant conferences. The trial’s outcomes will be made available to the public through a clinical trial registry like ClinicalTrials.gov, and addressed to the participants, their caregivers and the funding agencies.
Ethics statements
Patient consent for publication
References
Footnotes
Contributors All authors have met the ICMJE criteria for authorship by contributing substantially to the study’s conception, design, drafting and revision. BPDK, DB, SB-M and LDB conceived the study. M-MB, CA, DW, CD and FB contributed to the study design. BPDK, DB and AAD helped to prepare and test equipment critical for the preparation and conduct of the protocol. VW will conduct screening and coordination with the critical care team. Recruitment and consent will be done by BPDK and VW. CM and BPDK developed the analyses pipelines. BPDK, SB-M and LDB wrote the manuscript. All authors contributed to the revisions of the manuscript.
Funding This study is funded by the New Frontiers in Research fund (NFRFE-2021-00886). Doctoral training scholarship to BPDK is provided by the Fonds de recherche du Québec-Santé (BF2-313707) and Chaire Fondation Caroline Durand en traumatologie aigue de l’Université de Montréal.
Competing interests None declared.
Patient and public involvement Patients and/or the public were not involved in the design, or conduct, or reporting, or dissemination plans of this research.
Provenance and peer review Not commissioned; externally peer reviewed.