Article Text

Abstract
Introduction Sepsis is one of the most common risk factors for acute respiratory distress syndrome (ARDS). Neutrophil elastase (NE) is believed to be an important mediator of ARDS. When sepsis occurs, a large number of inflammatory factors are activated and released, which makes neutrophils migrate into the lung, eventually leading to the occurrence of ARDS. Sivelestat sodium is an NE inhibitor that can inhibit the inflammatory reaction during systemic inflammatory response syndrome and alleviate lung injury. Therefore, we hypothesise that intravenous sivelestat sodium may prevent the occurrence of ARDS in patients with sepsis.
Methods and analysis This is a prospective, investigator-initiated, double-blind, adaptive, multicentre, randomised, controlled clinical trial with an adaptive ‘sample size re-estimation’ design. Patients meeting the inclusion criteria who were transferred into the intensive care unit will be randomly assigned to receive sivelestat sodium or placebo for up to 7 days. The primary outcome is the development of ARDS within 7 days after randomisation. A total of 238 patients will be recruited based on a 15% decrease in the incidence of ARDS in the intervention group in this study. A predefined interim analysis will be performed to ensure that the calculation is reasonable after reaching 50% (120) of the planned sample size.
Ethics and dissemination The study protocol was approved by the Ethics Committee of ZhongDa Hospital affiliated to Southeast University (identifier: Clinical Ethical Approval No. 2021ZDSYLL153-P03). Results will be submitted for publication in peer-reviewed journals and presented at relevant conferences and meetings.
Trial registration number NCT04973670.
- Clinical Trial
- AUDIT
- Adult intensive & critical care
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Strengths and limitations of this study
This is one of the first prospective randomised controlled trials applying intravenous sivelestat sodium to acute respiratory distress syndrome in patients with sepsis.
It is a prospective, double-blind, adaptive multicentre, randomised controlled clinical trial with a re-estimation of the sample size.
Appropriate sample size calculation was not possible due to the lack of available high-quality clinical data; therefore, the sample size was calculated in two stages to ensure that the calculation was reasonable, maximising the possibility of obtaining significant results and providing credible outcome data.
As the duration and distribution of infected patients are unpredictable geographically and temporally, the number of recruited patients at each centre is also unpredictable, in spite of the competitive enrolment.
Introduction
Background and rationale
Acute respiratory distress syndrome (ARDS) is a life-threatening and critical illness syndrome that is characterised by alveolar capillary injury and hypoxemia, with overall mortality rates of 40%.1 Sepsis is one of the leading causes of ARDS,2 and up to 6.8–38% of patients with sepsis will develop ARDS.3 4 Despite advances in critical care management and lower tidal volume ventilation strategies,5–7 the treatment for sepsis-induced ARDS remains supportive, and the mortality is higher than that for sepsis alone.8 Therefore, how to prevent ARDS in patients with sepsis is of great significance.
In sepsis, the inflammatory storm produced in the body destroys the endothelial layer and induces endothelial cell leakage, and then neutrophils migrate into the alveoli9 and can release neutrophil extracellular traps and elastase. Neutrophil elastase (NE) can cause vascular endothelial injury, increase vascular permeability and the permeability of pulmonary endothelial cells and epithelial cells, cause protein and water in the plasma to leak out of the pulmonary vessels, cause alveolar oedema and bleeding and promote the occurrence and development of ARDS.9–11 It is one of the important pathogeneses of ARDS.
Studies have shown that sivelestat sodium, an inhibitor of NE, has obvious protective effects on hamster acute lung injury models caused by endotoxin, cobra toxin and hydrochloric acid inhaled from the trachea.12–14 Clinical studies have shown that sivelestat sodium can improve oxygenation, ameliorate lung injury15 and increase the time of ventilator weaning in patients with systemic inflammatory response syndrome-induced acute lung injury (ALI).16 However, it is unknown whether NE inhibitors can prevent ARDS in patients with sepsis.
The median time of onset of ARDS was 2 days after admission.17 The period from admission to the development of ARDS provides a short opportunity for the prevention of ARDS.3 Studies have shown that the release of NE in patients with high-risk ARDS increased significantly in the early stage.18 Therefore, it is of great clinical significance to prevent such patients from developing ARDS by inhibiting NE.
Thus, we hypothesise that intravenous sivelestat sodium in patients with sepsis within 24 hours after sepsis diagnosis might prevent the occurrence of ARDS. In this trial, we also will observe the effect of the drug on the improvement of oxygenation, 28-day ventilator-free days (VFD), 28-day time to clinical improvement, 28-day and 90-day mortality and so on.
Objectives
The main goal of this study is to determine whether sivelestat sodium has a protective effect on ARDS (Berlin definition)19 in patients with sepsis.
Methods and analysis
Design and setting
It is an investigator-initiated, double-blind, multicentre, prospective, randomised, controlled clinical trial with an adaptive ‘sample size re-estimation’ design.
This protocol was constructed in accordance with the Standard Protocol Items: Recommendations for Interventional Trials 2013 guidelines.20 The protocol is summarised in figure 1 and table 1.
Participant timeline

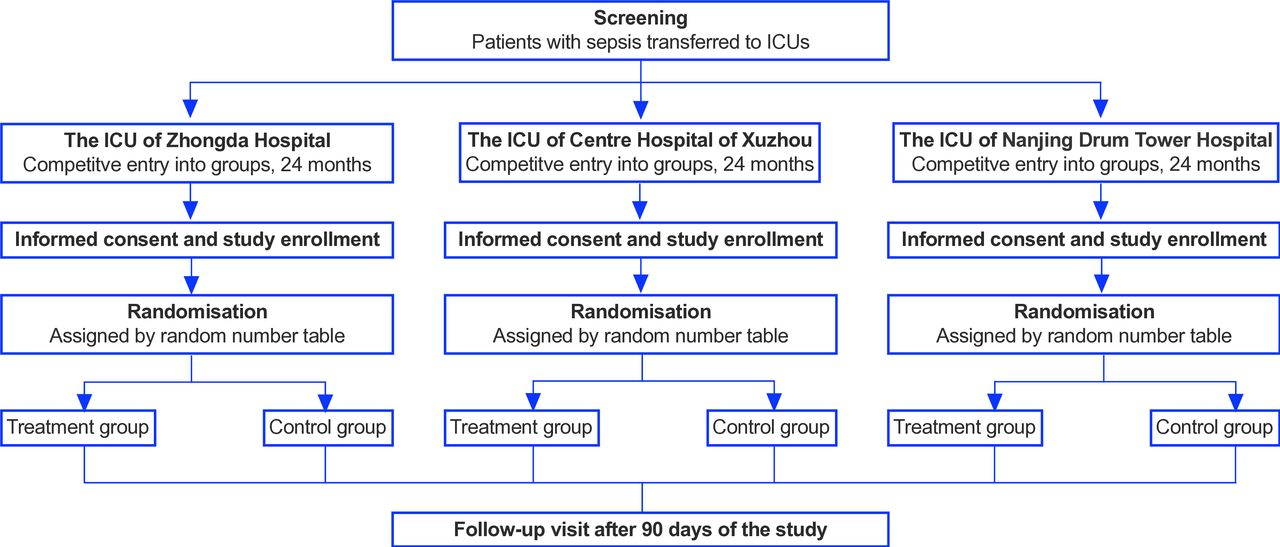{kind=link}
Study flow. ICU, intensive care unit.
Study sites and period
This clinical trial is being conducted in the intensive care unit (ICU) of Zhongda Hospital affiliated to Southeast University of Nanjing City, the ICU of Nanjing Drum Tower Hospital and Xuzhou Central Hospital from 1 October 2021. Zhongda Hospital affiliated to Southeast University is an affiliated hospital of medical teaching and research university, which is the leader of this study.
Study participants
The study will enrol ICU patients who were admitted to the above three hospitals with sepsis. The specific inclusion and exclusion criteria are as follows.
Inclusion criteria
Adults (age equal to or more than 18 years, equal to or less than 75 years old).
The sepsis 3.021 diagnostic criteria were met within 24 hours after admission.
The patients or their family members fully understood the purpose and significance of the trial, voluntarily participated in the clinical trial and signed the informed consent form.
Exclusion criteria
Patients identified with ARDS at the time of admission.
Patients who explicitly refused mechanical ventilation.
Patients with three or more extrapulmonary organ injuries and organ failure (single organ Sequential Organs Failure Assessment (SOFA) score ≥3).
Patients who need home oxygen therapy or with home mechanical ventilation (by tracheotomy or non-invasive ventilation, but excluding continuous positive airway pressure/bilevel positive airway pressure, only for patients with obstructive sleep apnoea).
Patients whose expected survival time is less than 48 hours.
Pregnant women and lactating women.
Patients with other conditions that were judged by the researcher to not be suitable for inclusion.
Study interventions
This study is an interventional study with two arms. The longest duration of the intervention group was 7 days. For the treatment group, 0.2 mg/kg/hour sivelestat sodium will be diluted in normal saline to a total volume of 48 mL and will be infused for 24 hours by an infusion pump. The control group will receive the same amount of normal saline containing only sivelestat sodium excipients. Except for the differences in the two drugs mentioned above, other critical supportive care such as anti-infection, fluid resuscitation and respiratory support were all carried out in accordance with international guidelines for the two groups. The intervention should be commenced within 1 hour after enrolment. Delivery of the assigned sivelestat sodium injection or placebo injection is supplied by Shanghai Huilun (Jiangsu) Pharmaceutical.
If the patient is transferred out of the ICU back to the general ward within 7 days, sivelestat sodium will be discontinued.
Risks, Adverse Events and Consent
As stated above, the trial is considered to be safe. First, the dose of sivelestat sodium has been demonstrated to be safe,16 even in patients older than 60 years.22 Second, some critically ill patients will be excluded based on the exclusion criteria. Third, the infusion rate is approximately 2 mL/hour, which is very slow. However, adverse events (AEs) and serious AEs (SAEs) must be observed and followed in accordance with the good clinical practice guidelines issued by the National Medical Products Administration of the People’s Republic of China.23
An AE refers to any untoward medical event that occurs after a human subject receives a drug. SAEs include prolonged hospital length of stay, disability, death and so on. AEs and SAEs will be recorded during the 90 days of observation from enrolment. Either may occur during a subject’s participation in the research and do not need to have a causal relationship with the treatment. The investigators will evaluate the relationship between the events and the intervention and will report it to the ethical committee and Data and Safety Monitoring Board (DSMB). Benefits and potential risks are written in the informed consent document. The patients will be informed of the purpose, intervention, benefits and possible risks of the study.
Who will take informed consent?
A member of the study team will take consent prior to the start of study activity. For patients who are unable to sign or initialise the consent form (online supplemental file 1), the consent form will be allowed to be signed and dated by his or her trustee or guardian.
Supplemental material
Additional consent provisions for collection and use of participant data and biological specimens
As a part of the consent form process, the participants will be required to provide authorisation for extraction and use of their data. The participants will also be asked for permission to gather de-identified information that may be used for ancillary studies. Biological samples obtained for further ancillary studies are not applicable in this study.
Randomisation, allocation concealment and blinding
The randomisation sequence will be created using SAS V.9.4 (procedure ‘PROC PLAN’) statistical software with a 1:1 allocation ratio using a random block size of 4. The participants, care providers, investigators, outcome assessors and statisticians will be blinded to the treatment allocation results. The sivelestat sodium and placebo were manufactured by Shanghai Huilun (Jiangsu) Pharmaceutical. All of them were identical in appearance and packaging.
Researchers should not unblind the investigational drug unless it is necessary to know the information of the investigational drug for medical treatment of the subjects. When a medical emergency occurs and it is necessary to immediately identify the type of medication taken by the subject, if possible, the relevant personnel of the main researcher and sponsor must be notified as much as possible before the trial drug is unblinded. The main researcher should conduct emergency unblinding to obtain specific grouping information of the subject. If the applicant is not contacted before unblinding, they must be contacted within 24 hours after unblinding. The researcher must record the unblinding date, location, reason, unblinding person, main investigator, relevant person in charge of the drug clinical trial institution, etc, and make detailed records in the case report form. Subjects who underwent emergency unblinding were treated as dropout cases.
Data collection and management
Baseline data, including demographic characteristics, admission diagnosis assigned group, Acute Physiology and Chronic Health Evaluation (APACHE) II scores, SOFA scores, Charlson comorbidities, source of infection and clinical information will be collected on the first day. The clinical information includes general vital signs, chest X-ray or CT, routine blood examination (red cell count, white cell count, neutrophil count, lymphocyte count, platelet count), blood biochemistry (alanine transaminase, aspartate aminotransferase, bilirubin and so on), coagulation function (prothrombin time, activated partial thrombin time, fibrinogen, D-dimer), cytokine tests (interleukin (IL)-1β, IL-6, IL-10 and so on), procalcitonin, C reactive protein (CRP), NE and blood gas analysis. The APACHE II scores, SOFA scores and clinical information will be collected on the 1st, 3rd and 7th days. The 28-day VFDs, shock-free days, length of hospital stay, time to clinical improvement, mortality and 90-day mortality will be evaluated during visits on the 28th day and 90th day.
Study outcomes
The primary outcome of this clinical trial is the development of ARDS, as defined by the Berlin criteria, within 7 days after randomisation.
The secondary outcomes include the following:
Oxygenation index (PaO2/FiO2) or SpO2/FiO2, concentration of inflammatory factors (including IL-1β, IL-6, IL-8, IL-10 and tumour necrosis factor (TNF)-α), concentration of NE, platelet count, concentration of CRP and SOFA score on days 1, 3 and 7 after drug administration.
The 28-day VFDs were defined as the number of days between successful weaning from mechanical ventilation and day 28 after study enrolment. For the patients ventilated for 28 days or more and for patients who died, the VFDs were 0.
Shock-free days (no vasopressor requirement) from day 1 to 28.
The 28-day time to clinical improvement24 was defined as the time from randomisation to an improvement of two points (from the status at randomisation) on a seven-category ordinal scale or live discharge from the hospital, whichever came first. The seven-category ordinal scale consisted of the following categories: (1) not hospitalised with resumption of normal activities; (2) not hospitalised, but unable to resume normal activities; (3) hospitalised, not requiring supplemental oxygen; (4) hospitalised, requiring supplemental oxygen; (5) hospitalised, requiring nasal high-flow oxygen therapy, non-invasive mechanical ventilation or both; (6) hospitalised, requiring extracorporeal membrane oxygenation (ECMO), invasive mechanical ventilation or both; and (7) death.
Length of hospital stay
The 28-day and 90-day mortality.
Participant timeline
The participant timeline is presented in table 1.
Sample size and interim analysis
The project statistician calculated the sample size based on the primary outcome before starting the trial. A sample size of approximately 238 patients is required to achieve 80% power at a 2.5% one-sided α level. On the basis of the results observed in the publication by Fein and Calalang-Colucci25 and Li et al,8 the incidence of ARDS in the control group was 18~38%. Based on these data, it is assumed that the incidence rate of placebo in patients with sepsis is 30% and that of treatment is approximately 15%.
The adequacy of sample size is crucial in clinical trials. Typically, the sample size is determined based on the results of one or more external pilot studies, butt appropriate sample size calculation was not possible due to the lack of available high-quality data. So, in our study, we are uncertain about the treatment effect of sivelestat sodium, which is expected to be around 15%. Given this uncertainty, sample size adjustment may be particularly important if the trial specifications have been made on preliminary and/or uncertain information. In our approach, we prospectively plan modifications to the sample size based on interim estimates of treatment effect for sivelestat sodium and placebo. An interim analysis will be performed when the recruitment rate reaches 50%. If strong evidence of effectiveness is observed with a p value<0.00258 (one-sided), the trial will be stopped early. Otherwise, if the conditional power (CP) is greater than 80%, the trial will continue as planned, and the sample size will remain unchanged. However, if the CP is less than 80%, we will re-estimate the sample size and increase it to ensure the statistical power of the trial.
The project statistician is responsible for authorising members of the Statistical Analysis Center (SAC) to access unblinded data and prepare reports for the DSMB. Members of the SAC will not be involved in the conduct of the study, other than preparation for and attendance at DSMB meetings. The sample size re-estimation will be conducted by an independent statistician based on the actual effect size in the interim analysis. The interim analyses lead to an inflation of the type I error, and the Lan-DeMets alpha spending function with an O’Brien-Fleming boundary was used to control type I error. If no more subjects are needed, early termination will be executed with the one-side 0.00258 nominal significance level.
Recruitment
Patient recruitment is currently being conducted in the three centres. The estimated recruitment in each centre is as follows: Zhongda Hospital, School of Medicine, Southeast University (80 participants); Nanjing Drum Tower Hospital (80 participants) and Xuzhou Central Hospital (80 participants). Recruitment started on 1 October 2021 in Zhongda Hospital. As of 31 March 2023, we had already enrolled 78 patients. There is no specific strategy to promote the rate of patient recruitment. Based on the results of the interim analysis, we will decide whether to terminate the trial or continue or add subcentres.
Statistical analysis
Statistical analysis will be performed using SAS V.9.4 statistical software. Continuous variables will be described using mean, SD, maximum, minimum, median, upper quartile (Q1) and lower quartile (Q3). Qualitative variables will be expressed using frequency and percentage. Two-sample t-tests will be employed for comparisons between the treatment groups when the data are normal and the Wilcoxon rank-sum test will be used for skewed distributions. X2 or Fisher’s tests will be used for categorical data and the Wilcoxon rank-sum test will be used for ordinal data. Significance tests will be two-tailed, with a statistical probability of p<0.05. The two-stage combination of primary outcome will be analysed by Cui-Hung-Wang (CHW).26
Primary and secondary analysis
The primary outcome, the incidence of ARDS, between the two study groups will be compared using the CHW method. The primary analysis of the primary endpoint will be performed without adjustment for baseline covariate imbalances using an intention-to-treat set and a per-protocol set. A sensitivity analysis of the primary endpoint will be performed after adjusting for baseline covariates, including sex, age, group assignment, source of infection and severity of illness. Last observation carried forwards imputation will be used for missing data. Planned exploratory subgroup analyses will be performed to investigate whether the treatment effect is modified by different initial baseline covariates.
Secondary binary outcomes between the two study groups, except for safety events, will also be compared using χ2 or Fisher’s tests. Two-sample t-tests or Wilcoxon rank-sum tests will be used for quantitative data. Safety analysis will be performed in a safety set, which is defined as a subset of subjects who were randomised and received at least one treatment.
Data safety monitoring
An independent DSMB comprised of two academic intensivists outside the study who are experienced in conducting clinical trials in critical illness is monitoring the progress and safety of the trial. The DSMB is able to pause the trial to investigate or give suggestions on any potential safety issues to improve the study design and implementation.
Patient and public involvement
There was no patient and/or the public involvement in the design, or conduct, or reporting, or dissemination plans of this protocol.
Ethics and dissemination
The study protocol was approved by the Ethics Committee of ZhongDa Hospital affiliated to Southeast University (identifier: Clinical Ethical Approval No. 2021ZDSYLL153-P03). Any changes in the study will generate synchronously protocol amendments, which will be submitted for approval to the Ethics Committee/institutional review board for filing in a timely manner. The changes will only be implemented after approval by the ethical committee. Once approved, the amendments will be circulated to the other study sites and ClinicalTrials.gov will be synchronously updated about any major changes.
This study results will be submitted for publication in peer-reviewed journals and presented at relevant conferences and meetings. The primary outcome of the study will be published as the first article and additional results derived from the data could be published in separate articles.
Discussion
Many recent studies have found that the elastase released by neutrophils is involved in the degradation of the main components of the extracellular matrix, a process that is closely related to lung injury.22 Inhibition of NE can improve the clinical outcome of patients with ALI, such as increasing the ventilator-weaning rate and ICU discharge rate.16
Sivelestat sodium is a selective NE inhibitor that can reduce pulmonary haemorrhage and exudation and reduce pulmonary oedema mainly by inhibiting the aggregation, adhesion and infiltration of neutrophils.9 It reduces the release of inflammatory factors such as IL-8 and TNF-α, inhibits inflammatory reactions and improves symptoms of lung injury.27 At present, sivelestat sodium has been widely used in the clinic, mainly for patients with ALI/ARDS.22 28 29
To the best of our knowledge, this is one of the first randomised controlled trials using sivelestat sodium to prevent the occurrence of ARDS in patients with sepsis. The treatment of ARDS is very limited, including low-tidal-volume ventilation, prone positioning, conservative fluid strategies and ECMO, but the effect is limited.30 ARDS brings a heavy medical burden to society.31 Therefore, this trial is of great significance, could save lives and allow for a lower economic burden. Sivelestat sodium is expected to inhibit sepsis inflammation, prevent ARDS and reduce mortality for patients with sepsis. In addition, it may also provide a basis and reference for the treatment of other diseases with similar mechanisms.
However, our study design also has some limitations. First, the upper age limit of this study is 75 years old. It is unknown whether patients with sepsis over 75 years old have the same result. Further research may be needed for these patients in the future. Second, we included patients with sepsis transferred to the ICU, and there may be selection bias for patients with sepsis not transferred in time. In addition, the patients with sepsis transferred from other hospitals may have had ARDS, and these patients were excluded from the study.
Trial status
The trial is currently ongoing at three sites (Zhongda Hospital, School of Medicine, Southeast University; Nanjing Drum Tower Hospital; and Xuzhou Central Hospital) in China. Enrolment started in October 2021 in Zhongda Hospital. Currently, all three sites are actively screening for patients. As of 31 March 2023, we had already enrolled 78 patients. The protocol version is 1.3 (12 October 2021).
Ethics statements
Patient consent for publication
Acknowledgments
The authors would like to thank the patients and their family members and the nurses, pharmacy staff, fellows and clinicians of the intensive care unit at the above hospitals for making this study possible. We would also like to express our gratitude to Shanghai Huilun Co. Ltd. for supplying sivelestat sodium and placebo.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors LL and YY are the principal investigators. SM, CL, ZG, JX, HQ, YY and LL designed the study protocol. SM wrote the manuscript. All authors contributed to revising the manuscript. The authors read and approved the final manuscript.
Funding This research is supported by the National Key R&D Program of China (No. 2022YFC2504405), the Clinical Science and Technology Specific Projects of Jiangsu Province (BE2020786), the National Natural Science Foundation of China (81870066, 82270083) and the Second Level Talents of the ‘333 High Level Talents Training Project’ in the sixth phase in Jiangsu (LGY2022025), Jiangsu Provincial Medical Key Laboratory (ZDXYS202205), Jiangsu Provincial Elderly Health Project (LD2021010) and Shanghai Huilun (Jiangsu) Pharmaceutical Co., Ltd.
Competing interests None declared.
Patient and public involvement Patients and/or the public were not involved in the design, or conduct, or reporting, or dissemination plans of this research.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.